Contents |
The current version of GARSA was implemented based on scripts, due to a tight schedule imposed by the need to obtain fast results on ongoing experiments. Next version is currently under development, and contemplates the use of Web services and parallelization techniques among others. Such development involves three master thesis and should be released within one year.
GARSAs architecture is supported by Perl scripts, resulting on some coding effort to add new tools to its pipeline. However, its implementation design is modular, well documented and based on development standards like page templates. Its flexibility has been proven by recent extensions, which have been developed by biologists with basic Perl knowledge, in short time.
The following options must be added to the the httpd.conf (Apache server) file:
<Directory "/var/www/html">
Options Indexes FollowSymLinks ExecCGI Multiviews
AddHandler cgi-script .cgi
AllowOverride AuthConfig
Order allow,deny
Allow from all
</Directory>
DirectoryIndex index.cgi
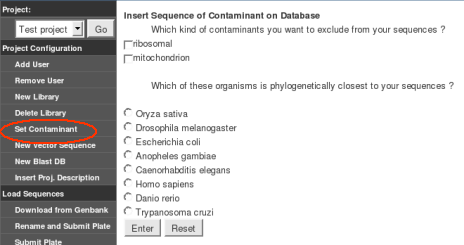
* Minimal requirements: GARSA is going to work with those (*) minimal packages, but in a limited way as similarity analyses won't be executed without NCBI-Blast and InterPro. Gene prediction won't be executed without Critica package, and Phylogeny without ClustalW and Phylip.
Download: Visit /garsa/ or contact Dr. Alberto Dávila (davila AT ioc.fiocruz.br)
For users interested in the GARSA platform, we offer the option to host their own projects on our servers, also providing advice and consultancy. At the present moment, we have 3 Intel Xeon Dual Processor servers and over 500GB of hard disk space available for this. Costs will be evaluated case-by-case. Users interested in this option should contact Dr. Alberto Dávila davila AT ioc.fiocruz.br.
The only way to create a new project in GARSA is having super-user privileges, either as admin_garsa or subadmin_garsa user. The admin_garsa user can grant subadmin_garsa privileges to several users, so they may create several new projects without the need to be the admin_garsa user.
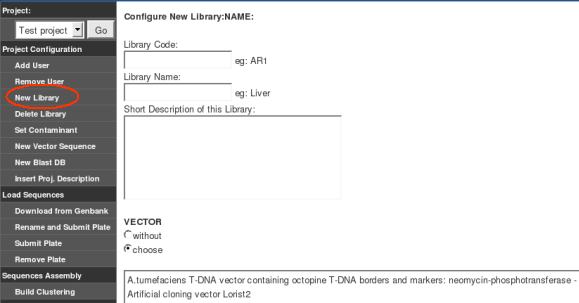
Any of the above mentioned users should use the "Create Project" option of the "Project Administration" menu to create a new project. GARSA ask for the following input data:
Project administrator login is created by GARSA based on "Project Code", eg: admin_TC, admin_DM or admin_PM
GARSA does not allow "admin_garsa" and "subadmin_garsa" users to manage projects, the only function assigned to these users is to create projects.
Once a new project has been created, GARSA will send all the details of the new project to the administrator's email.
When starting new libraries, GARSA asks for the following input data:
DMJS111001A07.g
DMJS111001C11.g
DMJS111001E08.g
In this case, resulting Zipped File should be named: DMJS111001.zip
DMJSABC100A07.b
DMJSABC100A07.b
DMJSABC100A07.b
And in this case: DMJSABC100.zip
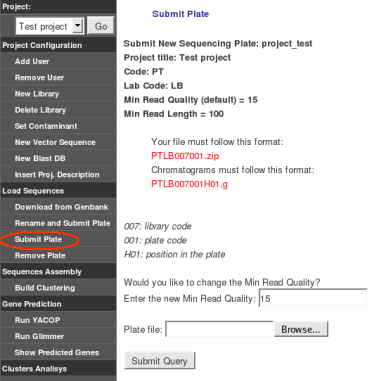
Only chromatograms from the same sequencing run or plate should be zipped together, resulting in a file as DMJS111001.zip or DMJSABC100.zip. This zipped file is the input for GARSA. Minimum read quality and minimum size length can be optionally modified here.
Each time a clusterization is done, GARSA produces 1 clusterization for each library plus 1 clusterization of all the libraries together. Only after clusterization has been done, GARSA allows project administrators to run Gene Prediction, Clusters Analysis and Sequence Annotation.
GARSA shows a warning message when users try to analyze non-clustered sequences:
GARSA can use Glimmer or the YACOP metatool (RBS, Critica, Zcurve) for gene prediction.
Glimmer needs (complete) CDS (multifasta format) of the organism under study or from a closely related species to be trained.
YACOP: Critica needs a set of nucleotide sequences from the organism under study or a closely related species. The nucleotide sequences needed by Critica must be formatted to be used by WU-Blast.
GARSA can use as many BLAST databases as your hard disk space can store. The New BLAST DB option allows the user to upload and format databases. TblastX, BlastX and BlastN options are active by default. However, only 2 BLAST runs are currently allowed to happen at the same time, in order to avoid CPU overload. E-value cut-off is configurable at this stage.
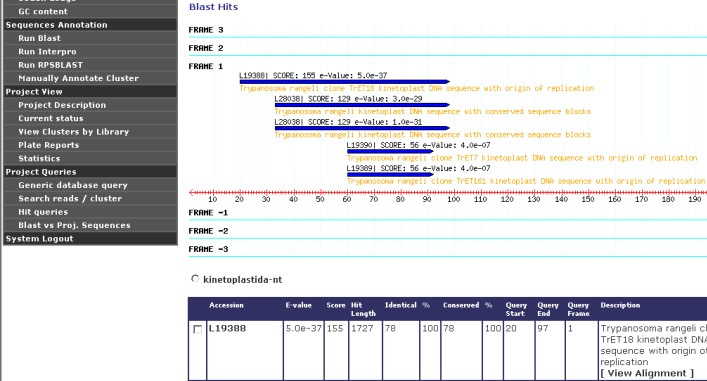
The following figure shows best BLAST results according to each frame shown, aiming to help with the identification of the right CDS frame:
The current version of GARSA works with InterPro 3.2, but the new version of GARSA (under development) will work with InterPro 4.0.
The Conserved Domain Databases from CDD, SMART, KOG, COG and KEGG are available.
Users can enter comments or notes for each cluter with this option. Notes entered one user cannot be deleted or modified by other users, allowing several users to work/comment the same clusters, in this manner sharing and complementing analysis.
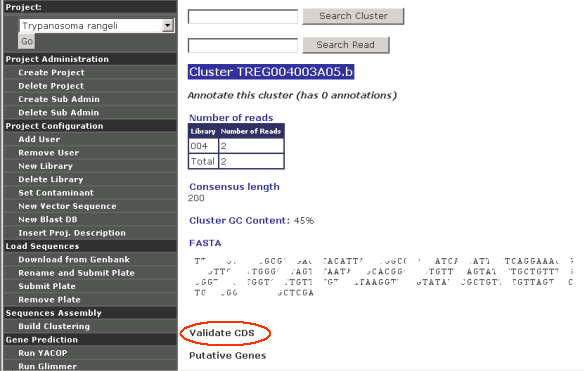
When a cluster is being viewed or examined, there is always a link to "Validate CDS":
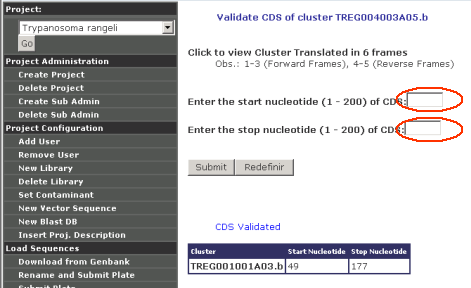
To Validate a CDS, users need to enter the beginning and end coordinates, then Garsa translates that sequence range using the TRANSEQ program of the EMBOSS package. Validated CDS always appear listed at the bottom of the page:
Generic database queries
A little console is presented, then users can query the MySQL database using MySQL command. For security reasons, only the SELECT command is allowed in this version.
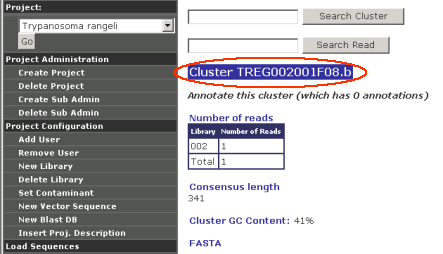
Search Reads/Clusters
A search tool to facilitate the finding of specific reads or clusters.
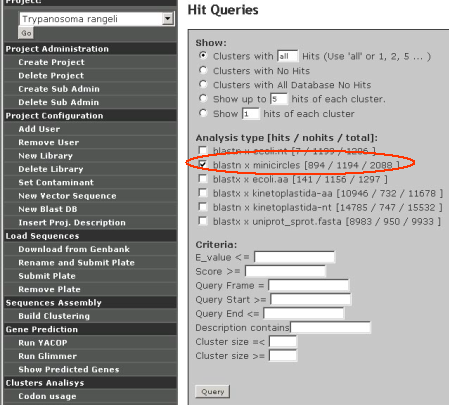
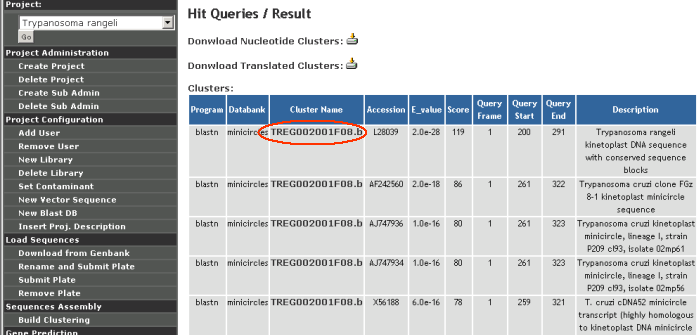
Hit queries
A number of options to query the different analysis results from GARSA. Clusters with a specific number of hits can be easily found. Clusters with no hits can be easily found with this feature.
Blast vs Project Sequences
Garsa uses "formatdb" from the Blast package to format "Reads" and "Clusters" to be used for WWW-Blast analysis, then any sequence can be query against "Reads" and "Clusters" of a given project in Garsa.
Users first need to clusterize sequences using "Build Clustering" in the "Sequence Assembly" menu. Most options from the menu are only available once sequences has been clusterized and BLAST done; those results are used to help with gene finding, alignment and phylogeny. For BLAST, "Run Blast" from the "Sequence Annotation" menu should be used. For Logo, users should first have Blast results (after clustering), then view results frrom a given cluster either via "View Clusters by Library" (Project View menu) or "Search reads / cluster" (Project Queries).
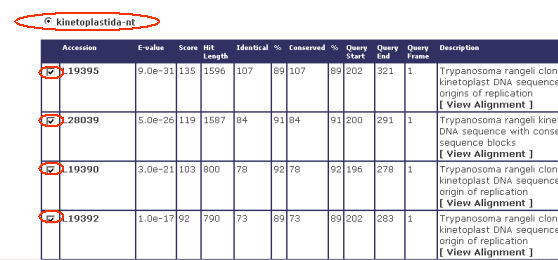
Once users are viewing BLAST results from a given cluster, they can select one of the BLAST DB's used (eg: kinetoplastida-nt) together with their respective results:
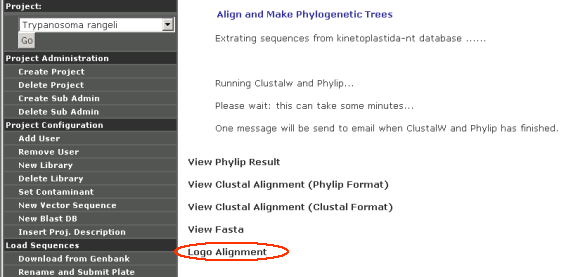
Select from the bottom option what type of sequences you want to analyze (eg: Nucleotide Sequences) then click "Run ClustalW and PHYLIP".
After that, ther user will be asked what substitution model that PHYLIP should use (eg., Kimura 2- parameter). Once the model is selected, the following screen will appear:
A first version of Garsa System documentation (still in portuguese) can be found at http://de9.ime.eb.br/~tgferreira/ip/Monografia_Vers%e3o1.0.pdf.
To Dr. José Marcos Ribeiro (NIAID/NIH) for suggestions and sharing his experience on EST analysis. To Dr. João Setubal (VBI and LBI/IC/UNICAMP) for allowing us to modify the algorithm for processing EST chromatograms. To MCT/CNPq, IAEA, CIRAD and FAPESP for financial support. To the Open Source Community for all the valuable help. To the authors of the softwares/modules used in GARSA for granting the academic and GPL licenses.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}